PIPELINE
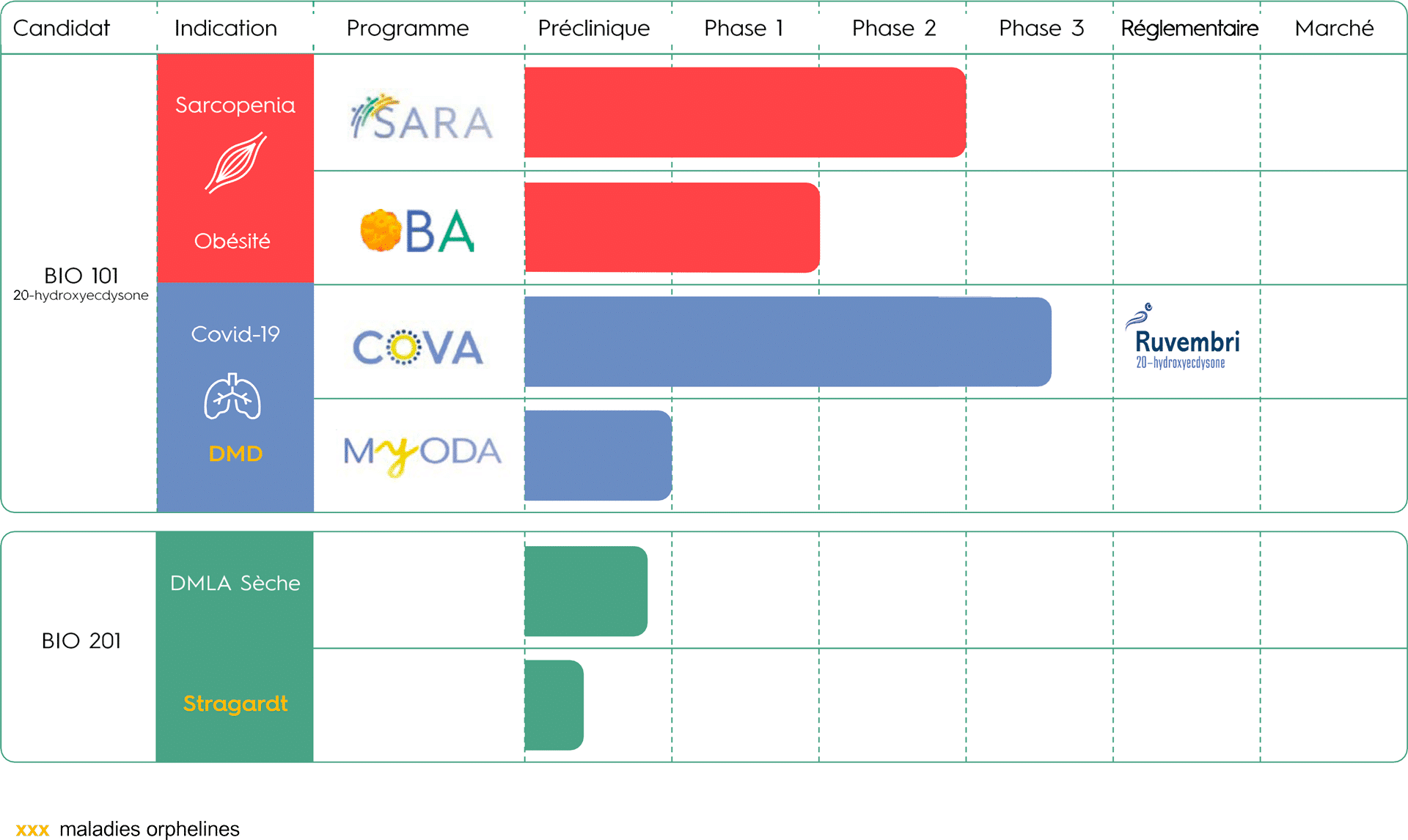
SARCOPÉNIE (PROGRAMME CLINIQUE SARA)
La sarcopénie est une dégénérescence des muscles squelettiques liée à l’âge, caractérisée par une perte de la fonction musculaire, de la force, de l’équilibre et de la capacité à se tenir debout et/ou à marcher. La sarcopénie conduit à une déficience motrice chez les personnes âgées (>65 ans) qui entraîne une perte d’indépendance et un accroissement des risques sur la santé, tels que des chutes, qui peuvent réduire l’espérance de vie.
La prévalence estimée chez les personnes âgées (≥ 60 ans) se situe entre 6 et 22%, une population qui devrait doubler, passant d’environ 962 millions d’habitants en 2017 à 2,1 milliards en 2050.
La sarcopénie a été décrite pour la première fois en 1989 et officiellement qualifiée de pathologie en 2016 par l’Organisation Mondiale de la Santé (OMS), qui l’a désignée sous le code ICD-10-CM code M62.84.
À l’heure actuelle, aucun médicament n’a été autorisé pour traiter la sarcopénie. Les recommandations actuelles en matière de traitement non médicamenteux sont principalement axées sur une activité physique modérée et une intervention nutritionnelle.
Nous avons testé l’innocuité et l’efficacité de BIO101 (20-hydroxyecdysone) dans le cadre d’un essai clinique de phase 2 mondial, randomisé, multicentrique, en double aveugle et contrôlé par placebo (SARA-INT) auprès de patients sarcopéniques présentant un risque de déficience motrice. Les Top Line Results (TLR) ont montré que BIO101 (20-hydroxyecdysone), à la dose la plus élevée (350 mg bid), permettait une amélioration cliniquement significative de 0,10 m/s au test de marche de 400 mètres (400MWT), le critère principal de l’étude. En outre, BIO101 (20-hydroxyecdysone) a montré un très bon profil de sécurité aux doses de 175 mg bid et de 350 mg bid, sans aucun événement indésirable grave (EI) lié au produit. Ces résultats prometteurs ont permis de définir les conditions du démarrage d’une étude de phase 3, encore jamais menée dans la sarcopénie.
Biophytis a obtenu l’autorisation de lancer l’étude SARA-31 en Belgique et aux États-Unis au cours du second semestre 2023. Le démarrage effectif de l’étude est planifié en 2024 et dépendra de la conclusion d’accords de partenariat et des moyens financiers de la Société.
OBESITÉ (PROGRAMME CLINIQUE OBA)
Dans le monde, environ 1 milliard de personnes sont atteintes d’obésité. L’obésité est une maladie chronique caractérisée par une quantité excessive de graisse corporelle qui perturbe le fonctionnement normal de l’organisme. Le traitement de l’obésité peut entraîner une perte de la masse et de la fonction musculaires, notamment en raison de régimes associés à un traitement basé sur les agonistes des récepteurs du GLP-1 récemment introduits sur le marché. Les agonistes des récepteurs du peptide 1 de type glucagon (GLP-1 RA) sont des médicaments très efficaces qui entraînent une perte de poids significative. Seulement, jusqu’à 40 % de la perte de poids totale provient des muscles, ce qui pose un réel problème car le tissu musculaire joue un rôle central dans le contrôle du métabolisme, en plus de sa fonction motrice.
BIO101 (20-hydroxyecdysone) est le premier activateur oral quotidien du récepteur MAS et a démontré des effets métaboliques sur les tissus musculaires et adipeux dans des études précliniques sur l’obésité. Ces effets bénéfiques de BIO101 (20-hydroxyecdysone) pourraient se traduire par une amélioration de la mobilité et de la force musculaire chez les patients obèses sarcopéniques, comme le suggère l’étude de phase 2 SARA-INT. Par ailleurs, la molécule 20-hydroxyecdysone a déjà été testée chez des patients obèses sous régime hypocalorique dans l’étude Quinolia, montrant des effets prometteurs sur la force musculaire et la perte de masse grasse.
Avec pour objectif de prouver l’efficacité de BIO101 (20-hydroxyecdysone) dans le traitement de l’obésité en combinaison avec les GLP-1 RA, l’étude clinique de phase 2 OBA devrait débuter mi 2024, avec des premiers patients traités au second semestre 2024. BIO101 (20-hydroxyecdysone) sera évalué chez des patients obèses traités par des GLP-1 AR et suivant un régime hypocalorique. Les premiers résultats de l’efficacité du candidat-médicament sont attendus pour 2025.
COVID-19 (PROGRAMME CLINIQUE COVA)
Environ 20% des patients atteints par le coronavirus 2019 (COVID-19) développent des formes sévères nécessitant une hospitalisation, parfois en service de soins intensifs. Le taux de mortalité de COVID-19 chez les patients âgés et/ou ayant des comorbidités sous-jacentes telles que l’hypertension, les maladies cardiovasculaires ou le diabète, est estimée entre 26 et 62%. Les formes sévères et leur évolution fatale sont associées à un syndrome de détresse respiratoire aigüe, une atteinte du myocarde, une dysfonction cardiaque, une arythmie et une altération de la fonction rénale. Du fait de son mécanisme d’entrée dans les cellules humaines, le coronavirus 2 du syndrome respiratoire aigu sévère (SRAS-CoV-2), responsable de la COVID-19, est notamment à l’origine d’un déséquilibre du système rénine angiotensine (SRA) associée à une inflammation, des phénomènes d’apoptoses et des coagulopathies.
Après la phase pandémique qui a eu un impact négatif considérable aux niveaux sanitaire, économique et social à l’échelle mondiale, la COVID-19 est désormais devenue une infection virale respiratoire endémique, au même titre que la grippe. Malgré les efforts considérables consentis en recherche, nous manquons toujours de traitements efficaces permettant de diminuer de façon significative la mortalité des patients atteints par les formes graves de COVID-19.
Nous pensons que la restauration de l’équilibre du SRA sera un moyen particulièrement efficace pour traiter les patients infectés par le SRAS-CoV-2. Ruvembri™ qui active le récepteur Mas, un élément clé du bras protecteur du SRA, permet d’améliorer significativement la fonction respiratoire dans différents modèles expérimentaux. Le programme clinique COVA évalue l’efficacité et l’innocuité de Ruvembri™ comme traitement pour prévenir la dégradation de la fonction respiratoire chez des patients ayant un syndrome de détresse respiratoire aigüe dû à la COVID-19.
L’étude de phase 2-3 COVA, portant sur des patients âgés de 45 ans et plus, hospitalisés pour des symptômes respiratoires sévères et présentant une infection prouvée par la COVID-19, a montré une réduction du risque d’insuffisance respiratoire ou de décès précoce à hauteur de 44%, statistiquement significative.
MYOPATHIE DE DUCHENNE (PROGRAMME MYODA)
La DMD est une maladie neuromusculaire génétique frappant les enfants de sexe masculin, caractérisée par une dégénérescence accélérée des muscles et responsable d’une perte de mobilité, de déficience respiratoire et de cardiomyopathie, conduisant à une mort prématurée. Il s’agit de la forme la plus courante de dystrophie musculaire chez les enfants, qui affecte environ un nouveau-né de sexe masculin sur 5 000 (environ 20 000 nouveaux cas par année dans le monde entier).
La DMD est provoquée par un certain nombre de mutations du gène de la dystrophine, qui entraînent l’absence ou de très faibles niveaux de dystrophine fonctionnelle, une protéine cytosquelettique qui protège les cellules musculaires.
L’absence de dystrophine dans les muscles affaiblit gravement la stabilité structurelle et membranaire des fibres musculaires entraînant la perte de la force, la mobilité ainsi qu’une altération de la fonction respiratoire et des déficiences cardiaques.
La progression de la maladie suit un cours extrêmement prévisible, typiquement décrit en 5 étapes consécutives : Pré-symptomatique, ambulant précoce, ambulant avancé, non ambulant précoce, non ambulant avancé.
Il n’existe actuellement aucun traitement pour la DMD et seulement des options thérapeutiques limitées, consistant principalement en corticostéroïdes et en deux thérapies ciblées (ciblant des mutations spécifiques de la dystrophine soit par saut d’exon, soit avec des codons stop) disponibles sur le marché (une aux États-Unis et une en Europe). Ces traitements ont pour but d’adresser la cause de la DMD en autorisant une expression partielle du gène de la dystrophine. Cependant, cette dernière produit une dystrophine tronquée qui ne permet pas la restauration complète de la structure cellulaire des muscles.
Dans les études précliniques animales, nous avons observé un effet positif sur la fonction musculaire, la mobilité et la fonction respiratoire (une invalidité majeure durant la phase ultérieure de la progression de la DMD) chez les modèles de souris mdx atteintes de DMD qui ont été traitées par BIO101 (20-hydroxyecdysone).
En 2018, nous avons obtenu de la U.S. Food and Drug Administration (FDA) et de l’Agence européenne des médicaments (EMA), la désignation de médicament orphelin pour BIO101 (20-hydroxyecdysone) pour le traitement de la DMD. La société prévoit de lancer une étude clinique de phase 1-2 (MYODA) au cours de l’année 2024.
DÉGÉNÉRESCENCE MACULAIRE LIÉE À L’ÂGE (PROGRAMME MACA)
La DMLA est une dégénérescence de la macula – la partie centrale de la rétine – liée à l’âge. C’est la principale cause de perte de vision irréversible et de cécité chez les personnes de plus de 60 ans. La dégénérescence maculaire liée à l’âge (DMLA) forme sèche affecte la vision centrale et perturbe différentes fonctions ayant un effet sur la qualité de vie et la capacité à vivre de façon indépendante, comme la lecture, la conduite automobile et la reconnaissance faciale. Il s’agit d’une maladie multifactorielle qui, selon nous, est principalement causée par l’accumulation d’A2E, un sous-produit du cycle des pigments visuels, qui entraîne une dégénérescence rétinienne.
Environ 85 à 90 % des patients atteints de DMLA présentent une forme de DMLA sèche, à un stade précoce, intermédiaire ou avancé, appelé atrophie géographique (AG). Il n’existe actuellement aucun traitement approuvé pour les différents stades de la DMLA sèche, y compris l’atrophie géographique.
Nous développons Macuneos (BIO201) pour traiter les patients atteints de DMLA sèche intermédiaire afin de prévenir l’évolution vers les stades avancés, la DMLA humide et l’AG, qui entraînent une perte de vision grave. Nous avons terminé les études de toxicologie animale chronique et aigüe nécessaires pour entamer les futurs essais cliniques.